


MolDesk Series
MolDesk (モルデスク)は、インシリコ創薬による、ドラッグデザイン(薬剤分子設計)・パッケージ・ソフトウェアです。計算エンジンとして、myPresto を使っています。PDB の任意の生体分子で GROMACS による MD 計算とトラジェクトリ解析 が試せます。
MolDesk Basic と MolDesk Screening があります。
- myPresto とは、AMED・ 経済産業省・NEDOの支援によって産業技術総合研究所・JBIC・大阪大学蛋白質研究所等によって開発されたJBICのソフトウェアです。最新版のソースコードはこちら (myPresto version 5 Download) からダウンロードできます。
- 弊社は、AMEDプロジェクト(ITを活用した革新的医薬品創出基盤技術開発 (平成25年度-29年度)、革新的中分子創薬技術の開発/中分子シミュレーション技術の開発 (平成30年度-令和2年度))に参加して、myPresto の開発と MolDesk を通して研究成果・技術の普及に努めております。
- MolDesk Basic または MolDesk Screening は、千葉工業大学(ひらめき☆ときめきサイエンス、高校生向け)、広島市立大学(ひろしま医工学スクール)、東京医科歯科大学、北海道大学、神戸大学、九州大学などの講義でも使われています。
- (株)バイオモデリングリサーチ様のたくさんのチュートリアル(日本語と英語)で、MolDesk のわかりやすい使い方が解説されています。
- myPresto の計算原理の解説は以下をご参考ください。動画「ドッキングソフトの原理と実際 1」「同 2」「同 3」そのテキスト、共立出版「タンパク質計算科学 基礎と創薬への応用 [CD-ROM付]」神谷 成敏, 肥後 順一, 福西 快文, 中村 春木 (著)
- CBI 学会 2020 年大会 企業セッションで、MolDesk の概要を説明しました。MolDesk および DataCheck サービス:myPresto システムの一例として
- 弊社社員が執筆者の一人の「創薬研究のための相互作用解析パーフェクト」羊土社の本は、実験から計算まで創薬研究に関連する幅広いテーマを扱っており、MolDeskについても説明があります。
低中分子化合物の創薬研究全体の流れを示すと共に、各テーマにおける重要なポイントがコンパクトに纏められている為、例えばドッキング・スクリーニング計算を行う際、使用する実験データの意味や背景をさっと確認するといった事にも利用出来ます。

販売実績
民間企業複数社、三重大学、北海道科学大学、安田女子大学、昭和薬科大学、名古屋大学、早稲田大学、奈良工業高等専門学校、崇城大学、岐阜大学、京都大学、大分大学、日本大学、NIH (National Institutes of Health)、慶応義塾大学、関西医科大学、近畿大学、立命館大学、明治薬科大学、国立環境研究所、広島市立大学、千葉工業大学、国立感染症研究所、電気通信大学、東京工業大学、北海道大学、産業技術総合研究所
MolDesk Screening ver.1.1.109 インシリコ スクリーニング 活性予測
MolDesk Screening マニュアルはこちらで公開しています。MolDesk Screening は、MolDesk Basic のすべての機能を含みます。
創薬向けに抽出した300万化合物ライブラリ(LigandBox 最新版をご提供)に対して、スレッド並列で高速にスクリーニングを行います。通常のPCでも、最大3日程度で高速に精度良くスクリーニング計算します(300万化合物の場合)。
ver.1.1.95 から Chiral 株式会社によるクラウドでの GROMACS 計算サービスが使えるようになりました。
ver.1.1.96 から Ab-initio 量子化学計算ソフト platypus-QM による RESP (RHF/6-31+G*) による化合物の電荷計算が可能になりました。
ver.1.1.83 から GROMACS による MD 計算とトラジェクトリ解析が可能になりました。 Windows版は、Windows 11 および Windows 10 ではユーザは GROMACS のインストールなしで GROMACS を使えます。Linux版、Mac版では ユーザが GROMACS をインストールする必要があります。Windows 10 で GROMACS 実行時の動画はこちら。
ver.1.1.71 回帰分析による化合物の各種物性特性値の予測が可能になりました。予測できる特性値は、LogP, LogS, LogD, Papp(膜透過係数), pKa などですが、ChEMBL から得られた実験データを使用して学習することが可能です。
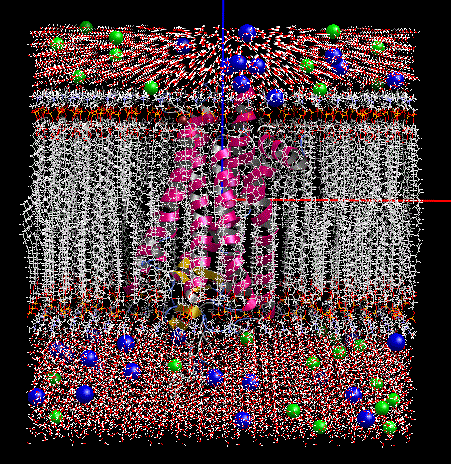
ver.1.1.70 膜貫通タンパク質を水溶液中の脂質2重膜に配置する MD 計算系を作成する機能で、6種類の脂質分子(DLPC, DMPC, DPPC, POPC, DOPC, POPE)を組成比で構成できるようになりました。MD 計算は、MolDesk Screening で動作する psygene で高速に実行できます。(系の作成は、MolDesk Basic、myPresto Portal でも可能です。)
ver.1.1.45 から docking score QSAR による化合物の活性値予測を実装しました。ChEMBL から得られた親和性(実験)データを使用して、特定タンパク質に対する化合物の活性値を予測します。
ver.1.1.39 から MVO Screening ( Maximum volume overlap method ) による類似構造探索を実装しました。
ver.1.1.19 から ユーザが用意したインハウス化合物ライブラリ(2次元 SDF 形式、数百~数百万分子)をスクリーニング対象とすることができます。
ver.1.1.18 から スクリーニング結果リストのクリックで3次元ドッキングポーズの表示が可能になりました。
MolDesk Basic ver.1.1.109 なんでもMD どこでもドッキング ポケット探索
MolDesk Basic マニュアルはこちらで公開しています。
通常、PDB や mmCIF などを入力したときに、各分子の力場の割り当てや、H原子の付加、RESP または MOPAC7 AM1-BCC による電荷付加、水溶媒の生成 などは非常に手間がかかります。数回のクリックで、力場 (AMBER ff99SB / AMBER GAFF2) を割り当てて、分子動力学計算とドッキング計算をシームレスに実行できます。中分子の化合物の AMBER GAFF2 による計算も簡単にできます。Molsite による高精度なポケット探索も行えます。
ver.1.1.95 から Chiral 株式会社によるクラウドでの GROMACS 計算サービスが使えるようになりました。
ver.1.1.96 から Ab-initio 量子化学計算ソフト platypus-QM による RESP (RHF/6-31+G*) による化合物の電荷計算が可能になりました。
ver.1.1.91 から Molsiteによる高精度なポケット探索が可能になりました。化合物の 3D化で並列処理を可能にして高速化しました。
ver.1.1.87 から 糖鎖分子を認識して SNFG表記で3D表示する機能が追加されました。
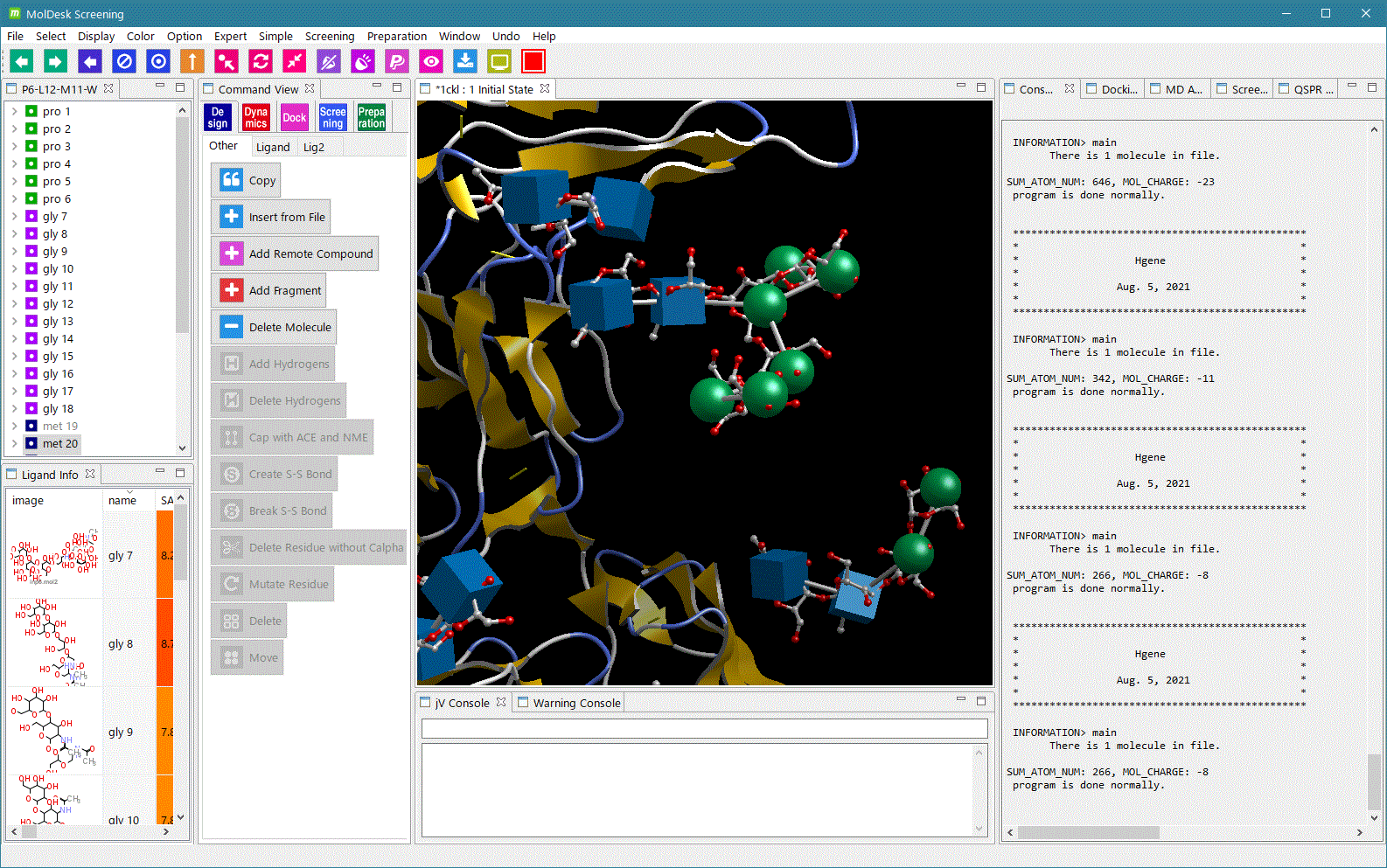
ver.1.1.83 から GROMACS による MD 計算とトラジェクトリ解析が可能になりました。 Windows版は、Windows 11 および Windows 10 ではユーザは GROMACS のインストールなしで GROMACS を使えます。Linux、Macでは ユーザが GROMACS をインストールする必要があります。Windows 10 で GROMACS 実行時の動画はこちら。
ver.1.1.78 から ドッキング計算で、核酸分子だけを対象にしたドッキング計算が可能になりました。
ver.1.1.66 から 数万個の分子を含む sdf / mol2 ファイルなどを高速に表示するビュアー機能を追加しました。
ver.1.1.62 から mmCIF ファイル(PDBx/mmCIF フォーマット)の入出力ができるようになりました。インターネット経由で入力するファイルは、これまでは PDB ファイルでしたが、今後は mmCIF ファイルベースになります。
※ 2019年7月1日より、結晶構造解析法によるPDBへの登録において、PDBx/mmCIFフォーマットファイルの登録が必須になることに対する対応です。
ver.1.1.39 から GROMACS のほぼすべてのトラジェクトリ解析結果の表示が、動画と連動して可能になりました。
ver.1.1.29 から 化合物の編集で、2Dエディター ( JChemPaint の最新版 ) が使用できるようになりました。2Dで編集した化合物は、ワンクリックで、3次元化して MolDesk に取り込めます。
ver.1.1.24 から myPresto (cosgene, psygene etc.) / DCD (CafeMol, MARBLE, CHARMM, NAMD etc.) / GROMACS / AMBER (netcdf 形式のみ) のトラジェクトリの動画や アニメーション GIF (animated GIF)ファイル出力が可能になりました。
ver.1.1.21 から PDB 入力の各分子鎖について、PDBj eF-site の正確な静電ポテンシャル面が表示できるようになりました。