MolDesk Screening の主な機能を説明します。
※ MolDesk Screening は、MolDesk Basic のすべての機能を含みます。
以下では、MolDesk Basic に含まれない機能を説明します。
最新版のマニュアルは、以下からダウンロードできます。
インシリコ薬剤スクリーニング [Screening]
ML-DSI / ML-MTS / MTS of myPresto
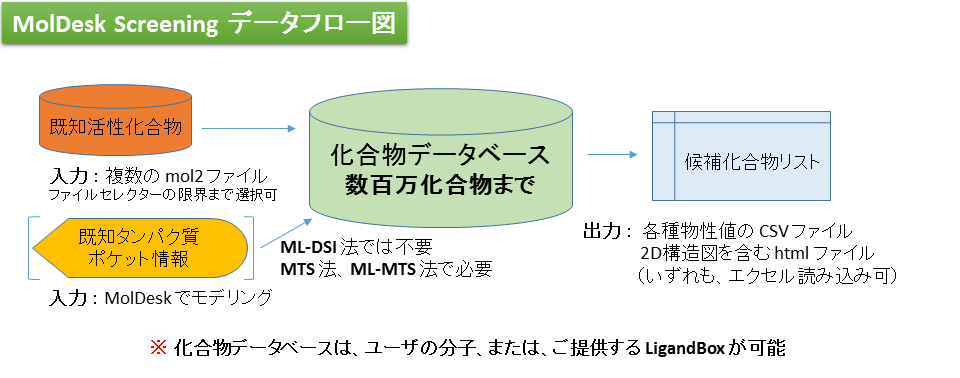
- 構造に基づく Virtual Screening (VS) のリランキング法として、myPresto が提供する Multiple Target Screening (MTS) 法および機械学習 MTS 法、リガンドに基づく VS のリランキング法として、同じく機械学習 Docking Score Index (DSI) 法 ※※ が利用できます。
- LigandBox ※ より創薬向けに抽出した300万化合物ライブラリに対して、スレッド並列で高速にスクリーニングを行います。
- ユーザが用意したインハウス化合物ライブラリ(2次元SDF形式、数百~数百万分子)をスクリーニング対象とすることができます。
- 各スクリーニング計算法の、化合物データベース以外の入力は以下の通りです。
| ターゲットタンパク質 (PDB) |
既知活性リガンド (MOL2) ※ |
|
| ドッキングスコア順 | ○必須 | |
| MTS | ○必須 | |
| 機械学習 MTS | ○必須 | ○必須 |
| 機械学習 DSI | ○必須 |
- SDF/MOL/SMILES ファイルは、MolDesk の機能によってあらかじめ MOL2 ファイルに変換する必要があります。
既知活性リガンドをスクリーニング対象に加えて、計算精度の検証ができます。計算精度は、Database enrichment曲線を表示してAUC(Area Under the Curve)によって確認します。
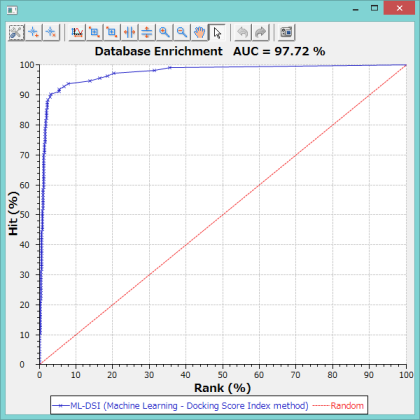
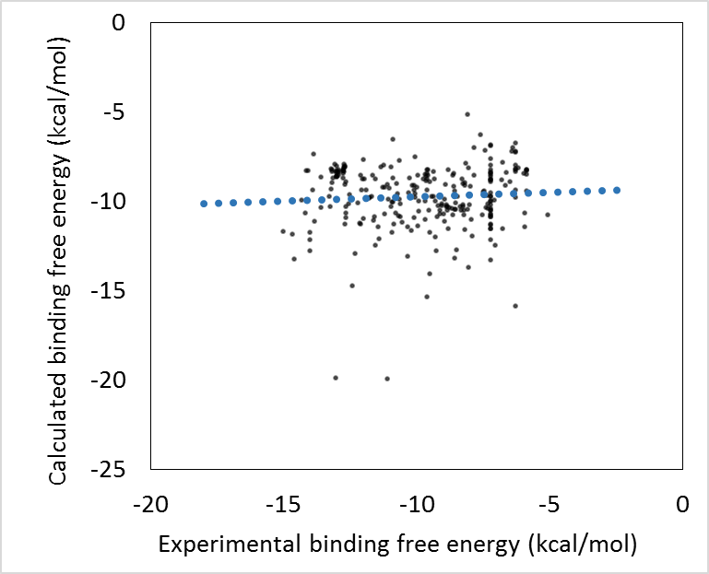
上記のグラフは、ターゲットタンパク質を用いない ML-DSI 法で精度の検証をした例です。AUC= 97.72% であることが確認されました。(シクロオキシゲナーゼ 4cox の活性化合物 124 個で機械学習して、113 個の別の活性化合物を LigandBoxの 10,000 化合物に混ぜて、スクリーニングした例)。
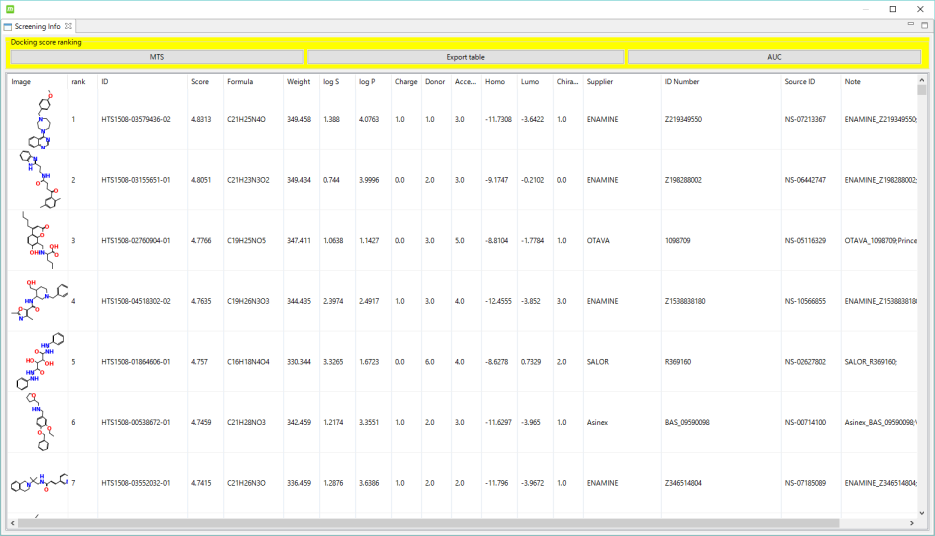
スクリーニング結果は指定リランキング順にリストされ、選択した化合物のドッキングポーズも確認できます。リストには化合物の 2 次元構造式、ランク、ID、スコア、化学式、分子量、LogS、LogP、電荷、ドナー数、アクセプター数、HOMO、LUMO、Chiral原子数、Source、Source ID が表示されます。各項目についてソート可能です。
リストを CSV または HTML 形式でファイル出力し、これらを Microsoft Excel に取り込んで化合物の発注に利用できます。
化合物データベースの作成
- インハウスの化合物ライブラリ( 2 次元 SDF 形式)を対象としてスクリーニングを行う場合、その前に各分子の立体構造を構築します。各分子の立体構造構築では、キラリティーを考慮した分子配座が生成されると共に、指定範囲外の分子量を持つ分子および不適当な構造を持つ分子のフィルタリングが行われます。
- 化合物-代表タンパク質で網羅的にドッキング計算をして相互作用行列を作成します。この計算はスレッド並列で行いますが時間がかかります。下記の表は、259,868 個の化合物の SDF ファイルを入力とした場合の計算時間です。実際の計算時間は、データベースを作成したい化合物数で比例倍してください。
| 通常のPC Windows 8.1 Intel Corei7-4790K 4.0GHz 16GBメモリ 8並列 |
計算サーバ Linux CentOS6 Intel Xeon(R) E5-2697 v2 @ 2.70GHz x 2 (24コア48論理プロセッサ) 64GBメモリ 48並列 |
|
| 259,868 個の化合物 | 641時間(26日7時間) | 191時間(7日23時間) |
並列計算による高速化
- スクリーニング計算は、化合物と代表タンパク質の全組み合わせで網羅的なドッキング計算をしますが並列計算で高速化します。
- 並列数は、Preference で設定できます。インストールしたPCや 計算サーバのプロセッサー数が、Preference の初期の並列数になってます。そのため、通常は、ユーザは何も特別な並列計算の設定をする必要がありません。
- 1回あたりのスクリーニングに必要な計算時間は以下の通りです。
| 通常のPC Windows 8.1 Intel Corei7-4790K 4.0GHz 16GBメモリ 8並列 |
計算サーバ Linux CentOS6 Intel Xeon(R) E5-2697 v2 @ 2.70GHz x 2 (24コア48論理プロセッサ) 64GBメモリ 48並列 |
|
| ドッキングスコア順 または、MTS |
35時間31分 | 10時間 3分 |
| ML-MTS | 45時間 7分 | 13時間12分 |
| ML-DSI | 8時間26分 | 2時間49分 |
タンパク質を含む受容体側が 8928 原子、LigandBox 200 万化合物 + 174 化合物、既知活性化合物 174 個 で計算した場合の例
化合物特性 [Screening]
活性予測 ( docking-score QSAR )
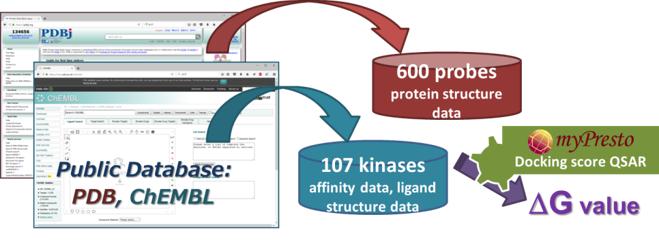
- myPresto の docking-score QSAR 法を利用して、化合物の結合自由エネルギー DG を予測します。Docking-score QSAR 法は、600 プローブ蛋白質に対するドッキングスコアを記述子として回帰分析を行い、化合物の DG を推算する手法です。
- 回帰に必要な DG およびプローブ蛋白質構造は、公的データベース(ChEMBL および PDB)より取得します。なお、DG は ChEMBL から得た親和性データ(IC50 値, % 阻害値, 活性値)から換算したものです。
- ChEMBL からダウンロードした特定のタンパク質に対する親和性データのファイルを、そのまま入力して計算できますので、実験データの加工をユーザはする必要がありません。異なる実験の親和性データの DG への変換や、化合物の 3次元化は、プログラム内部で自動的に行います。
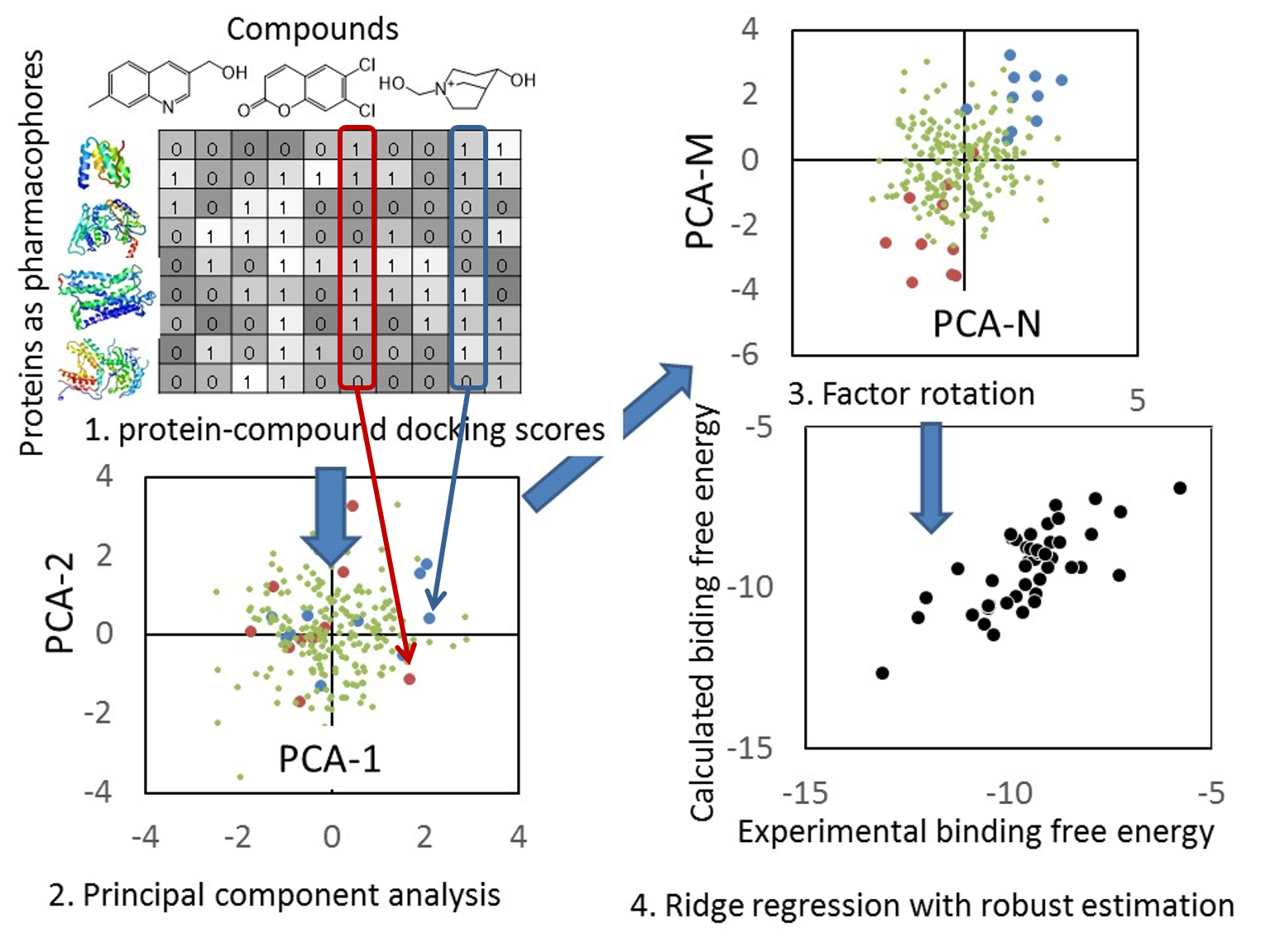
- 回帰モデルは、正規化項を伴った記述子ベースの重み付き PCR で構築しており、同時にロバスト推定(M 推定)を使用して、外れ値の影響を抑制しています。
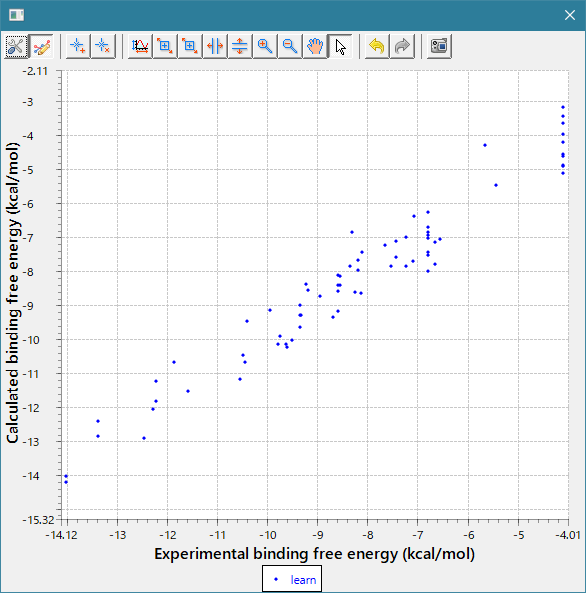
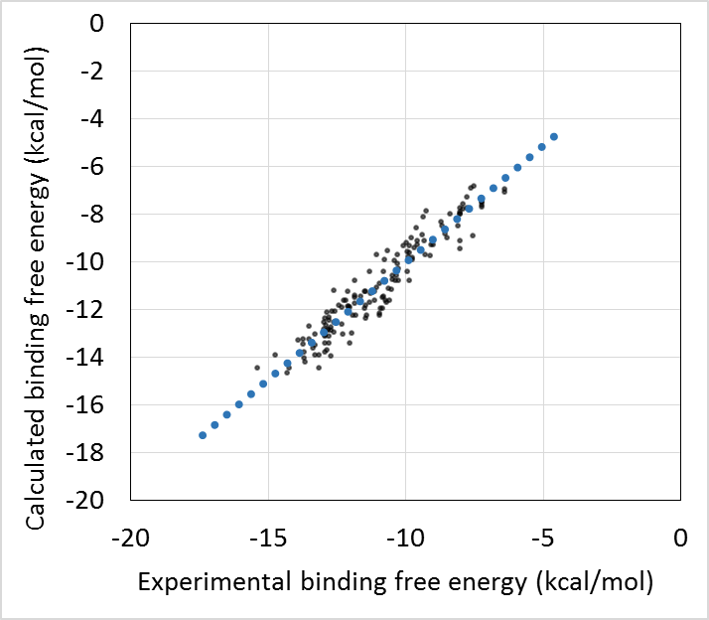
- 回帰モデル作成時の活性値の実験データと計算データの相関グラフをデフォルトで表示し、回帰モデルの精度を常に確認できます。(下図 上)
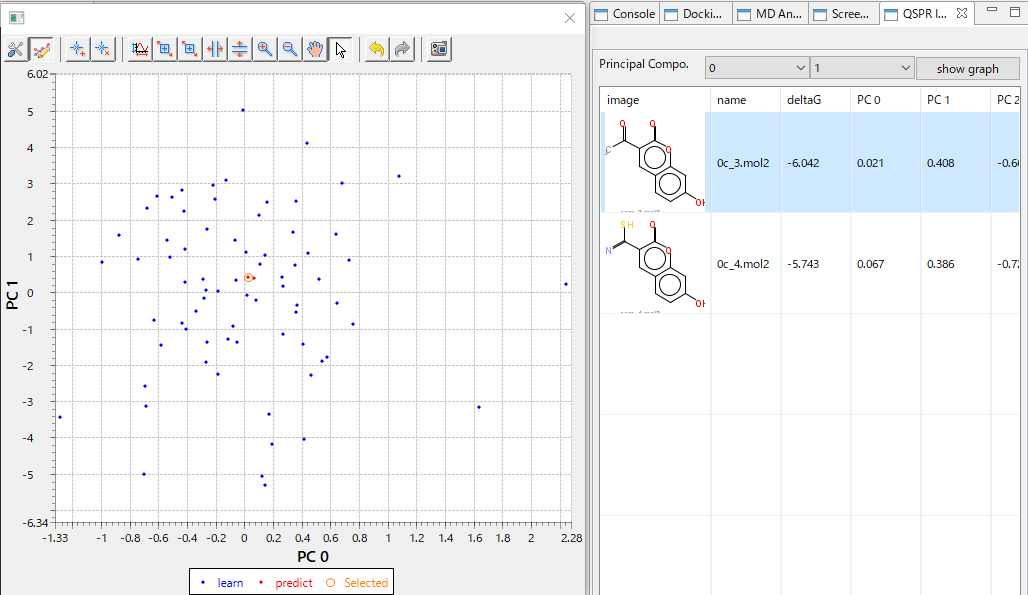
- 回帰モデルを使って活性値を予測したときの、PCA グラフを常に表示します。予測した化合物の PCA グラフ内での位置が確認できますので、ChEMBL 由来の化合物の集団から予測化合物がかけ離れてないかで、予測の信頼性を評価できます。(下図 下)
- ChEMBL の実験データを入力にして回帰モデルを作成する工程では、ファーマコフォアを代表した 600 種類のタンパク質に対するドッキング計算を実行するため、比較的長時間の計算時間が必要です。このため、回帰モデル作成の工程を、弊社で請け負うことも可能です。
MVO Screening ( Maximum volume overlap method )
- MVO Screening とは、MIN-MVO (energy MINimization Maximum Volume Overlap) ※ を利用してクエリ化合物に似た構造を持つ化合物をスクリーニングします。
- MIN-MVO 法は、クエリ分子と DB 分子の重ね合わせを行い、体積の重なり度合い(%) をスコア計算すると共に、併せて分子配座を計算する手法です。
- 入力として 1 分子以上のテンプレートを必要としますが、蛋白質のポケット内で体積重ね合わせと配座探索を energy minimization によって同時に行うことができる為、上のスクリーニング法に比べて精度の向上が期待できます。
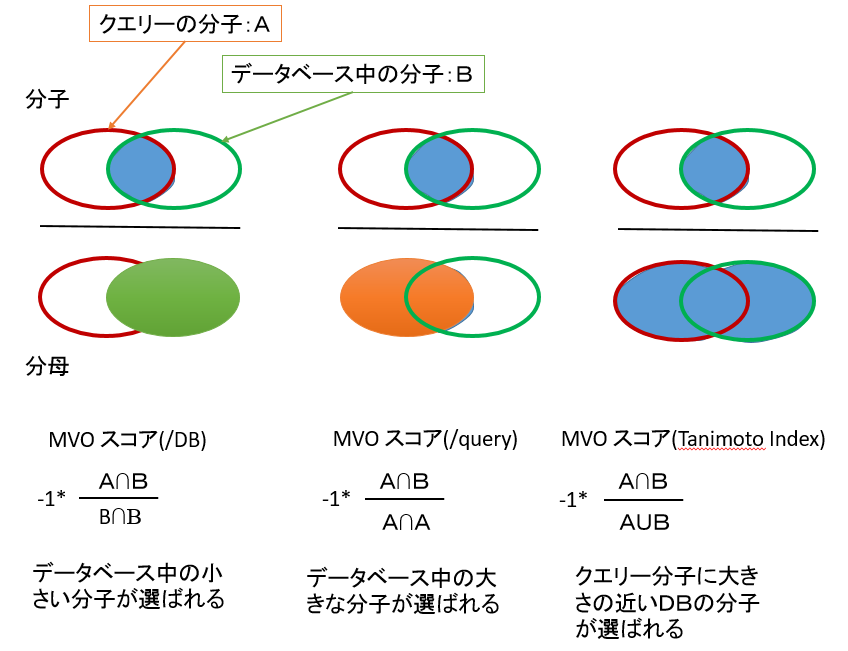
- 重ね合わせた体積の指標から、/DB・/Query・-1*Tanimoto の 3 種類の MVO スコア計算することにより、それぞれ出力傾向の異なるリストを得ることができます。各スコアの得られるリストの傾向および定義は、下の通りです。
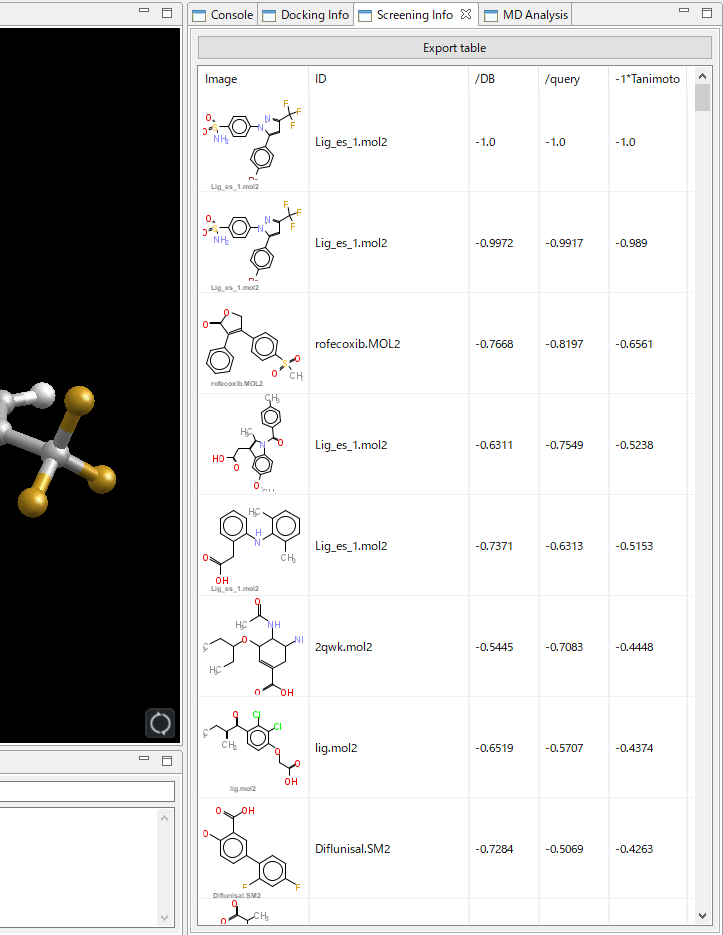
検索結果リスト例
類似構造検索
- TGS 法 (Topology Graph Similarity ※) による化合物分子の類似構造検索をします。
- 系の中の化合物を1分子選択します。この分子が検索のクエリーになります。ファイルセレクタ―によって、検索される側の複数の分子を選択して、検索します。
- 検索される側の複数の分子は、あらかじめ mol2 ファイルをユーザが用意します。
- 検索結果は、順位、mol2 ファイル名、TGS スコアを、以下の通りリスト表示します。TGS スコアは、同一化合物の場合に 0.0 となり、スコアが大きくなると類似性が低くなります。
部分構造検索
Substructure Search ※
- 系の中の化合物を1分子選択します。この分子が検索のクエリーになります。ファイルセレクタ―によって、検索される側の複数の分子を選択して、検索します。
- 検索される側の複数の分子は、あらかじめ mol2 ファイルをユーザが用意します。
- 検索結果は、内部番号、mol2 ファイル名、原子数、見つかった部分構造の数を、以下の通りリスト表示します。
文献リストとやや詳しい解説
スクリーニング (MTS / DSI)
- MTS (Multiple Target Screening) 法とは、標的蛋白質構造をもとにした structure-based のインシリコスクリーニング法であり、DSI 法とは、既知活性物質をもとにした ligand-based のインシリコスクリー ニング法です。ヒット探索のための in silico スクリーニングは、通常、標的蛋白質に対して、化合物ライブラリーに含まれる多数の化合物を順次ドッキングし、スコアの良い化合物をヒット化合物候補として採択します。しかし、このような一般的に用いられる手法では、ヒット化合物の予測精度は、ランダムスクリーニングよりは良いが低いままです。MTS 法、DSI 法では、多数の蛋白質と化合物ライブラリーを準備し、蛋白質-化合物相互作用行列をあらかじめ作成して計算に用いることで、ヒット化合物の予測率を向上しています。
Multiple target screening method for robust and accurate in silico screening.
Journal of Molecular Graphics and Modelling.25, 61-70. (2005)
Y. Fukunishi, Y. Mikami, S. Kubota, H. NakamuraImprovement of protein-compound docking scores by using amino-acid sequence similarities of proteins.
Journal of chemical information and modeling.48, 148-156. (2008)
Y. Fukunishi, H. NakamuraClassification of chemical compounds by protein-compound docking for use in designing a focused library.
Journal of Medicinal Chemistry. 49, 523-533. (2007)
Y. Fukunishi, Y. Mikami, K. Takedomi, M. Yamanouchi, H. Shima, H. NakamuraFinding ligands for G-protein coupled receptors based on the protein-compound affinity matrix.
Journal of Molecular Graphics and Modelling.25, 633-43. (2007)
Y. Fukunishi, S. Kubota, H. NakamuraAn efficient in silico screening method based on the protein-compound affinity matrix and its application to the design of a focused library for cytochrome P450 (CYP) ligands.
Journal of chemical information and modeling. 46, 2610-22. (2006)
Y. Fukunishi, S. Hojo, H.NakamuraNoise reduction method for molecular interaction energy: application to in silico drug screening and in silico target protein screening.
Journal of Chemical Information and Modeling. 46, 2071-2084. (2006)
Y. Fukunishi, S. Kubota, H. NakamuraA virtual active compound produced from the negative image of a ligand-binding pocket, and its application to in-silico drug screening.
Journal of Computer-Aided Mol Design. 20, 237-48. (2006)
Y. Fukunishi, S. Kubota, C. Kanai, H. Nakamura
LigandBox (化合物データベース)
- LigandBox は、世界中の2次元の SDF 形式の電子カタログを集め、H 原子の付加、3次元構造化、原子電荷や各種特性値も付加して DB 化したものです。類似構造検索により重複を除き、最終的には 300万化合物に集約しています。具体的には、H 原子の付加は、水素の解離状態は水中での主たるイオン形で行い、3次元構造化は、AMBER/GAFF2 力場で構造最適化し、電荷の付加は、MOPAC 7 AM1 モデルでの Mulliken population で行っています。SDF に基づく異性体を考慮し、ファイル形式は、Sybyl mol2 です。
配布する LigandBox には各種特性値を記述した化合物の mol2ファイルの他に、スクリーニング計算に必要な 化合物-代表タンパク質のドッキング計算によって得られた相互作用行列も含みますので、myPresto の MTS, ML-MTS, ML-DSI 法でのスクリーニングができます。
LigandBox: A database for 3D structures of chemical compounds.
BIOPHYSICS. 9, 113-121 (2013).
T. Kawabata, Y. Sugihara, Y. Fukunishi, and H. Nakamura.
LigandBox でヒットした化合物の発注方法について
LigandBox は、ナミキ商事(株)様、キシダ化学(株)様よりご提供頂いた化合物リストから化合物ライブラリを構築しています。両商社を経由した発注手順は以下の通りです。
- 入手試薬のリストアップ(各試薬の量・純度を指定)
- 各商社の化合物IDで試薬リスト案を作成、見積りを依頼
- 各商社からの見積り回答を基に試薬リスト案を調整
試薬プロバイダーの在庫状況・所在国による輸出入規制などにより、供給可能な試薬が異なります。また、各商社による法規制調査の結果、代替品の提案が行われる場合、輸送の問題等で納期に間に合わない場合もあり、それらを勘案して試薬リスト案の調整を行います。 - 試薬リストの決定
調整を反映して決定した優先順位に基づき、在庫率に応じて少し多めに試薬リストを作成します。また、試薬が足りない場合の取り決め等を行います。 - 見積・発注・納品
この様に各商社と対話的に作成した試薬リストに基づいて、試薬の正式な見積り・発注・納品となります。
Docking score QSAR
- Docking score QSAR は、多数の蛋白質に対するドッキングスコアの重み付き平均で結合自由エネルギーを推算する方法です。推算モデルは、リッジ回帰を用いた記述子ベースの重み付き主成分解析 (PCA) で計算しており、ロバスト推定(M推定)を利用して外れ値の除外を行って精度を良くしています。
- 公共データベースの活用
回帰に利用する親和性データおよび構造データは、ChEMBL および PDB より取得します。ChEMBL より得られた親和性データ ( IC50 値, % 阻害値, 活性値など) は、全て結合自由エネルギー DG に換算します。但し、ChEMBL には換算に必要な実験情報が不足していた為、幾つかの仮定を置いています( Kd = Ki 等 )。ChEMBL の様な公的なデータベースを基にしつつも、相関係数 0.87(誤差 0.92 kcal/mol)で化合物の活性予測ができます (molprof 内部データによる)。
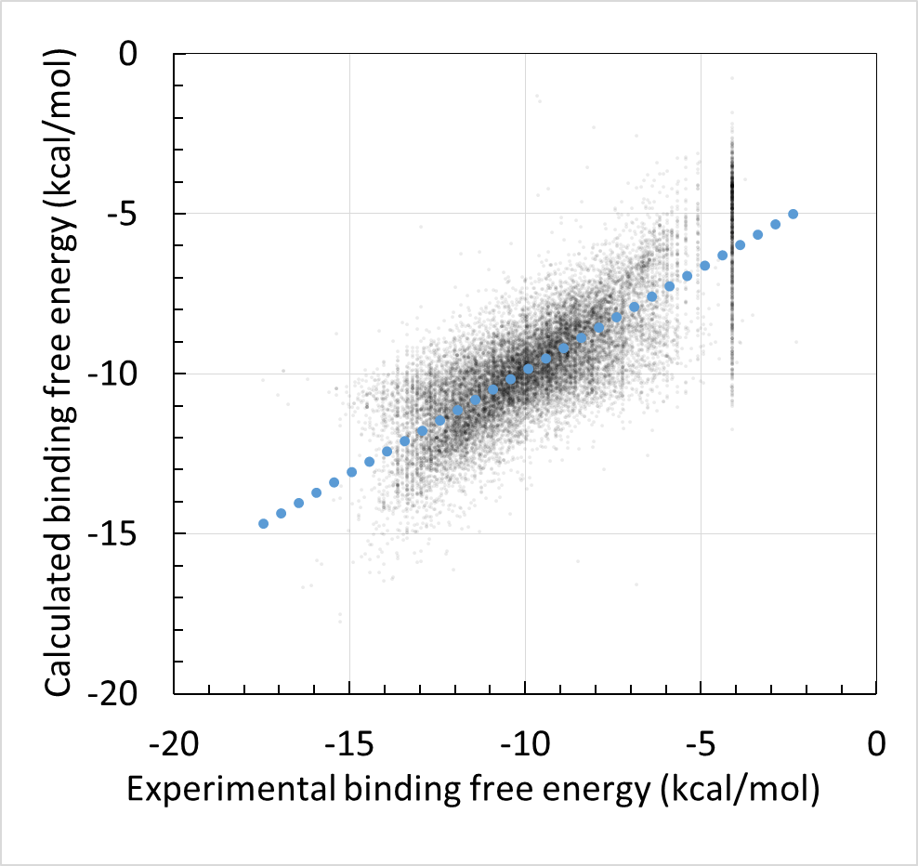
- MMP (matrix metalloproteinase) に対する適用
MMP は結合ポケットに Zn イオン( Zn2+ )を含んでいる為、結合エネルギーの評価が難しいです。この MMP についても、Docking score QSAR は良好に動作し、docking score = DG と見なした単純な推算結果に対して精度の大幅な向上が見られました。

Prediction of Protein−compound Binding Energies from Known Activity Data: Docking-score-based Method and its Applications
Mol. Inf. doi:10.1002/minf.201700120 (2018 Feb.)
Yoshifumi Fukunishi, Yasunobu Yamashita, Tadaaki Mashimo, and Haruki NakamuraQuantitative Structure‐activity Relationship (QSAR) Models for Docking Score Correction
Mol. Inf. 2017, 36, 1600013.
Yoshifumi Fukunishi, Satoshi Yamasaki, Isao Yasumatsu, Koh Takeuchi, Takashi Kurosawa, and Haruki Nakamura
MVO Screening
- MVO Screening (旧名:MD-MVO (molecular dynamics maximum volume overlap method))は、化合物の 3 次元構造を基にして、類似の 3 次元構造を持つ化合物を探索する手法です。2つの分子の体積の重なりを原子電荷の類似性も考慮して行い、体積の重なり具合をスコアとします。分子の重ね合わせ計算では、エネルギー極小化計算を用い、分子の配座、変形も考慮して重ね合わせができるため、既知化合物の類似化合物探索では高いヒット率を示します。
A new method for in-silico drug screening and similarity search using molecular-dynamics maximum-volume overlap (MD-MVO) method.
Journal of Molecular Graphics and Modelling. 27. 628-636. (2009)
Y. Fukunishi, H. Nakamura
類似構造検索
- Topology Graph Similarity は、分子の共有結合をエッジとした分子グラフを、エッジ行列表示とし、その行列固有値を指標とし、化合物の類似性を探索する手法です。分子の構造情報は、実数値のベクトルへと変換され、ベクトルの距離から類似性が計算されます。非常に高速ですが、光学異性体、配座を区別することはできません。
A similarity search using molecular topological graphs.
Journal of Biomedicine and Biotechnology. Article ID 231780. (2009)
Y. Fukunishi, H. Nakamura
部分構造検索
- Substructure Search は、複数の検索対象化合物 (mol2 形式) に対し、クエリ構造 (mol2 形式) と同じ構造を持つ化合物を検索します。分子は化学結合をエッジとするエッジ行列に変換され、部分構造の比較は、ウルマンの定理によって行います。分子の配座、光学異性体は考慮することができません。